Modern biological, translational and biomedical research is powered by massively parallel assays that provide tens of thousands, hundreds of thousands, and indeed now with Next Generation Sequencing, billions of measurements at a time from a biological or medical sample. Over ten years ago, I embarked on a career that was focused on the optimization of methods for the analysis of such data. being skeptical of the naive application of standard biostatistical methods in high dimensions, and being aware that different methods would lead to different conclusions about the genome and its function, the path I chose was the empirical, objective comparative evaluation of alternative methods for data analysis. This involved the use of real and simulated data to hold regular 'bake-offs' to compare methods. We have acquired an immense amount of practical knowledge, including improvements in how to think about evaluating alternative methods for data analysis. This type of methodological research has required the development of objective measures by which to compare the performance of methods and algorithms that implement such methods. Ultimately, this search led to a paradigm for complex data analysis I call Intelligently Informed Empirically Optimized Data Analysis, which involves analyzing a given data set only after determining which, among many alternative methods, might be optimal for that particular data set.
Virtually all of data analysis for the -omic sciences follows a similar path, after data generation:
Data -> Data Representation -> Data Normalization -> Interpretative Analysis
Data
The data may be, continuous qualitative or semi-quantitative measurements (such as global gene expression microarray data), or they may be discrete (as in variants in Next Generation Sequence data). They may be a mix of genetic, genomic, proteomic data with clinical co-variates and clinical outcomes. The most frequent types of studies are case/control or treatment/control studies,
Data Representation
However it is collected, it is often changed by some 1st-level 'analysis'; perhaps background noise in subtracted from the data (should it be?); perhaps the data are log-transformed (should they be?); perhaps the data are normalized among samples (should they be, if so how?); perhaps they are filtered using some data quality measure (should they be, if so, which method for assessing data quality, and which threshold?).
All of these cognate questions are questions about just the basic measurements to be analyzed, and yet other than a general gestalt 'feeling' about the distribution of the data, it seems that no paradigm for data analysis has been developed that has systematized a process to inform the data analyst on these questions in an objective manner.
Data Normalization
Systematic variation among measurements from the same sample (sample-level bias) can usually be addressed via mathematical manipulation (such as dividing all measurements by a global mean; or subtracting from each measurement from a sample the difference between the global mean or median for the sample and the global mean or median from the entire data set). The cognate questions then become (1) do the data need normalization, and (2) if so, how (by which method)?
Interpretative Analysis
In this step, the actual 'statistical' analysis might be done, to identify genes with significant differences in expression between or among groups; to identify alleles with significantly different frequencies between or among sample groups; to identify putative biomarkers that indicate clinical outcome, for example. Most of the time, this type of analysis is done in a univariate sense. Prevailing practices (i.e., 'habit') often determines which univariate test might be used. However, given a sample size, is that univariate test warranted? Is it sufficiently powerful? Or maybe we should just take ratio of means between two groups (no, I'll show why not later in another post). Maybe we should take the difference without bothering with statistical inference, and follow-up our study with an attempt to validate the potentially interesting leads?
Maybe we should use more than one criterion for finding potentially interesting leads in our data (consensus)?
So far, just in these steps, I have exploded method space. If P is the number of way data can be pre-treated (i.e., filtered or background-'corrected'), and R is the number of ways data can be scaled, and N is the number of ways data can be normalized, and T is the number of 'tests' or measures of difference or association, the method space of M methods that exists is at least
M = P x R x N x T (1)
I say at least M because very often there are parameters within options at each stage (stringency of filtering; intensity of normalization) that can also add significantly to the method space. Also, filtering for data quality and back-ground 'correction' are actually separate issues. Moreover, the scaling transformations can be done in combination (e.g., log2 followed by z-transformation), and tests can be used in various logical combination (union, intersection; etc).
M = 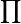 Pi x Rj x Nk x Tp (2)
Pi x Rj x Nk x Tp (2)
Pi x Rj x Nk x Tp (2)
Those of us involved in bioinformatics data analysis are fully aware
that alternative combinations of methods options can yield completely
different sets of results. This provides substantial motivation for
careful consideration of methodological alternatives, using the paradigm
of Intelligently Informed Empirically Optimized Data Analysis.
Enumeration of these methods for various applications is simple;
creating software that is capable of executing various combinations of
methods and providing objective evaluation measures is challenging, but
it has also proven an extremely worthwhile endeavor.
The number of conceivable possible ways to analyze the data is immense (2); however, which combination, for each data analysis instance, is optimal, can be determined with sufficient focused effort on careful, unbiased evaluation using objective measures.
These measures can include internal measurement consistency of replicates (measurement reproducibility); internal reproducibility of the inference; and, somewhat naively, some measures of low measurement variance across biological replicates (groupwise global correlations; coefficients of variation). Among these, the one that has proven most reliable, and intuitive to biologists, is internal reproducibility of the inference.
Software can be built that implements all M over a range of parameter
space, providing critical and useful objective feedback to the data
analyst on which methods might be used, and which might be avoided due
to the undesirable influences on the properties of the data and the
ensuing inferences.
In future posts, II will provide information on and examples of some of the gems of strategies of high-dimensional -omic data analysis that we have learned in hopes of enabling others to improve their research via better informed data analysis, and provide some examples of how we use these principles over many data sets to inform ourselves on the utility of various means for data analysis in genome and proteome-scale dimensions.
Future Posts
How to Be Smart About Next Generation Sequence Data Analysis
log2(PM-MM): A Terribly Expensive Lesson in -Omic Data Representation
Why We Should Avoid Analyzing Log-Transformed Data in High Dimensions
Why So-Called 'Fold-Change' Absolutely Lies in High and Low Dimensions